Гипотеза развития эссенциальной гипертонии
1. Гипотеза Э. Гелльгорна и соавт.
Инициальным патогенетическим фактором является стойкая повышенная возбудимость и реактивность (гиперергия) высших симпатических нервных центров (в частности, расположенных в заднем отделе гипоталамуса). Факторами, вызывающими стойкую гипе-рергию этих центров, является длительное, повторное возбуждение эмоциональных центров, тесно связанных с симпатическими ядрами гипоталамуса.
Повышение тонуса прессорных (симпатических) центров ведет к спазму сосудов, увеличению сердечного выброса и повышению АД, обусловливает гиперпродукцию гуморальных факторов с прессорным действием адреналина, норадреналина, вазопрессина,
АКТГ, кортикостероидов, а также гиперсекрецию ренина в юкстагломерулярном аппарате (ЮГА) почек.
2, Гипотеза Е. Муирад, А. Гайтона и соавт.
Инициальным фактором развития гипертензии является генетически обусловленный пониженный уровень ЫаС1- и водовыделительной функции почек, что ведет к накоплению избытка натрия и воды в организме, включая ткань сосудистых стенок, в том числе их гладкие мыпщы. В связи с этим развивается гиперволемия, повышаются тонус сосудов и чувствительность их стенок к прессорным гормонам и другим биологически активным факторам.
3, Гипотеза нарушения функций мембранных ионных насосов проф. Ю.В. Постнова.
Инициальным фактором в патогенезе артериальной гипертензии является генерализованный наследственный дефект мембранных ионных насосов клеток, включая глад» комышечные клетки стенок артериол. Дефект этот заключается в снижении активности кальциевого насоса, локализующегося в мембранах эндоплазматической сети, и натриевого насоса, локализующегося в плазмолемме. В результате уменьшается «откачивание» Са+2 из цитоплазмы в эндоплазматическую сесть и Иа+ из цитоплазмы в межклеточное пространство. Избыток Са+2 и Ыа+ в цитоплазме гладкомышечных клеток сосудов вызывает их спазм, а также повышение чувствительности к прессорным факторам.
4, Гипотеза проф. Г Ф. Лонга и проф. А.Л. Мясникова.
Инициальным патогенетическим фактором развития ГБ является растормажива-ние центров ССС (снижение тормозного влияния коры головного мозга на подкорковые вегетативные нервные центры, прежде всего пресеорные).
Этиологическими факторами являются стрессоры. Первичное звено патогенеза -возбуждение симпатических центров -> включение еимпатоадреналовой реакции, подключение ЮГА почек -» альдостерон -» накопление Иа»1″ -» увеличение ОЦК + набухание сосудистой стенки .*-» повышение ее реактивности. Длительный спазм гладких мышц сосудов -> накопление Са+2 в цитоплазме мышечных волокон -» активация РНК-полимеразы -> гипертрофия гладкомышечных элементов сосудистой стенки.
Клинико-потегенетинеские формы эссенциальной гипертонии[по Кушаковско-му].
1. Гиперадренерическая (Ланг, Мясников).
2. Гипергидратационная.
3. Ангиотензин-зависимая.
4. Кальций-зависимая (Постнов, Орлов).
5. Цереброишемичеекая.
В табл. 3.7.3. и 3.7.4. представлены прогностические факторы артериальной гипертензии (стратификация факторов риска).
Стратификация риска ГБ позволяет врачу составить более объективное (хотя и упрощенное) представление о долговременном прогнозе заболевания у каждого конкретного больного с целью выбора оптимальной тактики индивидуального лечения.
А. Факторы риска сердечнососудистых заболеваний
В. Поражение органов-мишеней
С. Сопутствующие клинические состояния
I. Используемые для оценки риска
Уровни систолического и диастоличе-ского АД (артериальная гипертензия 1-3 степени)
Мужчины старше 55 лет
Женщины старше 65 лет
Курение
Уровень сыворотки общего холестерина более 6,5 ммоль/л (250 мг/дл)
Сахарный диабет
Указания на преждевременное развитие сердечно-сосудистого заболевания в семейном анамнезе
И. Другие факторы, оказывающие неблагоприятное влияние на прогноз
Пониженные уровни холестерина ЛПВП
Повышенные уровни холестерина ЛПНП
Микроальбуминурия (30-300 мг/сут) при сахарном диабете
Нарушенная толерантность к глюкозе
Ожирение
Сидячий образ жизни
Повышенные уровни фибриногена
Социально-экономическая группа с высоким риском
Этническая группа с высоким риском
Географический регион с высоким риском
Гипертрофия левого желудочка (по данным электрокардиографии, эхокардио-графии или рентгенографии органов грудной клетки)
Протеинурия (>300 мг/сут) и/или небольшое повышение концентрации креати-нина в плазме (1,2-2,0 мг/дл)
Ультразвуковые или рентге-ноангиографнческие признаки атеросклеротического поражения сонных, подвздошных и бедренных артерий, аорты
Генерализованное или фокальное еужение артерий сетчатки
Сосудистое заболевание головного мозга
Ишемический инсульт
Геморрагический инсульт
Преходящее нарушение мозгового кровообращения
Заболевание сердца
Инфаркт миокарда
Стенокардия
Реваскуляризация коронарных артерий
Застойная сердечная недостаточность
Заболевание почек
Диабетическая нефропатия
Почечная недостаточность (содержание креатинина в плазме крови выше 2,0 мг/дл)
Сосудистое заболевание
Расслаивающая аневризма
Поражение артерий с клиническими проявлениями
Выраженная гипертоническая ретинопатия
Кровоизлияния или экссудаты
Отек соска зрительного нерва
Примечание. Поражения органов-мишеней (ПОМ) соответствуют II стадии гипертонической болезни по классификации экспертов ВОЗ 1996 г., а сопутствующие клинические состояния — III стадии заболевания.
| Таблица 3.7.4. Уровень риска сердечно-сосудистых осложнений у больных с артериальной гипертензией разной степени с целью определения прогноза* [по ВОЗ-МОГ, 1999]. | |||
| Факторы риска (кроме артериальной гипертензии) и анамнез болезни | Уровень риска при артериальной гипертензии | ||
| 1-я степень («мягкая») | 2-я степень («умеренная») | 3-я степень («тяжелая») | |
| : Нет других факторов риска | Низкий | Средний | Высокий |
| 1-2 других фактора риска | Средний | Средний | Очень высокий |
| 3 и более других факторов риска, ПОМ или сахарный диабет | Высокий | Высокий | Очень высокий |
| Сопутствующее заболевание* * | Очень высокий | Очень высокий | Очень высокий |
* Типичные примеры риска развития мозгового инсульта или инфаркта за 10 лет: низкий риск — менее 15%; средний риск — примерно 15-20%; высокий риск — примерно 20-30%; очень высокий риск — 30% или выше. ** См. табл. 3.7.3. ПОМ — поражение органов-мишеней (см. табл. 3/7.3Л.
Порочные круги при гипертонической болезни:
1. Рениновый: повышение АД ~» артериальный спазм -> ишемия почек -* выброс ренина из ЮГА -> активация РААС -> повышение АД.
2. Сердечный: гиперволемия ~» закон Анрепа (сила сердечных сокращений пропорциональна давлению (сопротивлению), против которого она работает) г-» рост УО ~» повышение АД.
3. Атеросклероз вызывает поражение депрессорных зон.
4. Сосудистый: спазм гладких мышц сосудов -» накопление Са+2 в цитоплазме мышечных волокон -> активация РНК-полимеразы -» гипертрофия и гиперплазия гладкомы-шечных элементов сосудистой стенки.-» повышение периферического сосудистого сопротивления.
///. Симптоматические гипертензии.
Симптоматические (вторичные) артериальные гипертонии (гипертензии)—
стойкое повышение АД, являющееся проявлением (симптомом) какого-либо заболевания.
1. Почечные артериальные гипертензии:
■ Вжоренолъная: атеросклероз почечной артерии, фибромускулярная дисплазия, аор-тоартериит, васкулиты, эндартериит, тромбоз, эмболия, аневризмы почечной артерии, атрезия и гипоплазия почечных артерий, артериовенозные фистулы, стенозы и тромбозы вен, гематомы, травмы сосудов почек; неоплазмы, сдавливающие почечные артерии -> ишемия почек -» выброс ренина из ЮГА -» активация РААС -> повышение АД.
■ Реноприеная: поражение (гломерулонефрит, хронический пиелонефрит, диабетический гломерулосклероз, амилоидоз, гидронефроз) или удаление паренхимы почек -> дефицит почечных простагландинов А и Е -> повышение АД.
2. Эндокринные артериальные гипертензии:
■Минералокортикоидные (первичный альдостеронизм или синдром Конна): гипер
плазия или опухоль клубочковой зоны коры надпочечников -> повышение продук
ции альдостерона -» усиление реабсорбции Ка+ и воды -» увеличение концентра
ции Ка+ в плазме -» гиперосмия плазмы -» стимуляция секреции вазопрессина ->
рост ОЦК -> повышение АД.
Увеличение концентрации Ка* в плазме -» повышение транспорта ионов Иа* через мембраны клеток -» накопление избытка Ка+ в клетках -» набухание клеток, в том
числе и стенок сосудов -» сужение их просвета + увеличение чувствительности к катехоламинам, вазопресеину, АГИ-» повьппение АД.
■ Глюкокортикоидные: гиперплазия или гормонально-активные опухоли пучковой зоны коры надпочечников (синдром Иценко-Кушинга) и базофильных клеток передней доли гипофиза (болезнь Иценко-Кушинга) .-> пермиссивное действие глю-кокортикоидов на эффект адреналина + слабая минералокортикоидная активность глюкокортикоидов -» повьппение АД.
■ Катехоламиновые: феохромоцитома (опухоль мозгового вещества надпочечников) -» гиперпродукция адреналина и норадреналина -> повышение АД.
■ Гипертиреоидные: тиреотоксикоз -» гиперпродукция тироксина и трийодтиронина -» увеличение силы и частоты сердечных сокращений (прямое кардиостимули-рующее действие гормонов + увеличение чувствительности сердца к катехолами-нам)^ увеличение УО -> повышение АД.
■ Вазопрессиновые: повышение секреции гипоталамусом вазопрессина (вазопресси-нома) -» сужение сосудов + задержка воды с увеличением ОЦК -» повышение АД.
3. Нейрогенные артериальные гипертензииформируются при органических поражениях нервных структур (сосудистые, воспалительные заболевания, опухоли ЦНС, травмы мозга), регулирующих системную гемодинамику (нервы, продолговатый, спинной мозг, гипоталамус, кора больших полушарий) -» преобладание прессорных влияний над депрес-сорными.
4. Гемодинамические артериальные гипертензиивозникают при поражении сердца и крупных сосудов:
■ Атеросклероз аорты -> поражение делрессорных зон аорты -> повьппение АД.
■ Коарктация аорты -> по закону Анрепа (сила сердечных сокращений пропорциональна давлению (сопротивлению), против которого она работает) -> рост УО -» повышение АД.
■ Недостаточность аортальных клапанов -> рост УО по закону Франка-Старлинга (чем больше наполняется сердце во время диастолы, тем сильнее оно сокращается в систолу) -» повьппение АД.
■ Полная атриовентрикулярная блокада.
■ Реологическая гипертензия: увеличение вязкости крови (болезнь Вакеза, полици-темия, эритроцитозы, гиперпротеинэмия) -> повышение АД.
■ Васкулиты (аорто-артериит или болезнь Такаясу) -> уменьшение диаметра сосудов -> повышение АД.
Источник
В соответствии с нейрогенной теорией патогенеза (Ланг Г. Ф., 1950; А. Л. Мясников,1965) артериальная гипертензия относится к болезням нарушенной регуляции АД. Авторами был собран обширный клинический материал, свидетельствующий о роли психоэмоционального «перенапряжения» в нарушении деятельности высших нервных центров, регулирующих сосудистый тонус. Перенапряжение и срыв корковых нервных процессов возбуждения и торможения приводит к образованию корково-подкоркового комплекса (доминанты возбуждения) с вовлечением в него адренергических структур заднего гипоталамуса, ретикулярной формации, что приводит к повышению симпатического тонуса и активации ГГНС. А. Л. Мясников высказал гипотезу о том, что дисфункция прессорных центров головного мозга возникает вследствие расстройства их трофики. Действительно, ограничение кровоснабжения, ишемия головного мозга приводят к подъему АД.
Поскольку психоэмоциональное перенапряжение, как правило, приводит к увеличению продукции и выброса во внутреннюю среду организма КА, тесно связанной с этой теорией можно считать и концепцию повышения тонуса СНС как ведущего механизма развития артериальной гипертензии. Причины, приводящие к повышению активности симпатического отдела ВНС, могут быть различными. Это, помимо нарушения центральных механизмов регуляции кровообращения, изменение чувствительности барорецепторов, нарушение метаболизма НА в синаптической щели, увеличение количества и (или) повышение чувствительности адренорецепторов на мембране гладкомышечных клеток сосудов, собственно мембранные нарушения сосудистого эндотелия, усиливающие их констрикторный ответ и др.
Еще одна современная концепция патогенеза артериальной гипертензии связана с нарушениями обмена ионов кальция в регуляции АД. Это теория «патологии клеточных мембран»(Ю. В. Постнова и С. Н. Орлова, 1987). Суть этой теории заключается в том, что у больных первичной артериальной гипертензией имеется генетически обусловленный дефект клеточных мембран, вызывающий нарушение трансмембранного переноса моновалентных катионов и накопления в цитоплазме кальция. Следствием этого является повышение содержания кальция в нейронах симпатических ганглиев, гладкомышечных клетках сосудов, кардиомиоцитах. Это приводит к усилению симпатических влияний и повышению чувствительности гладкомышечных элементов сосудов к прессорным стимулам.
Ряд концепций патогенеза рассматривают повышенное АД как закрепленную компенсаторную реакцию, направленную на сниженную перфузию определенных тканей. Еще-в 1940-х гг. Н. Н. Савицкий высказывал предположение, что артериальная гипертензия является способом компенсации нарушения кровотока в жизненно важных органах.
Согласно церебро-ишемической теории (Диккинсон, 1965 г.) уменьшение общего мозгового кровотока является инициальным звеном в генезе эссенциальной гипертензии. В соответствии с этой теорией, атеросклероз или длительный спазм магистральных артерий мозга ведет к дефициту кровоснабжения, вследствие чего происходит активация механизмов, направленных на нормализацию мозгового кровотока, в частности, СНС, что приводит к возрастанию АД и компенсаторному увеличению притока крови к мозгу.
К теориям нарушения регуляторных механизмов АД, как причине развития артериальной гипертензии, относят теорию «переключения почки»(Гайтон, 1987 г.). Основой этой концепции являются экспериментальные и клинические данные, демонстрирующие развитие гипертензии на фоне гиперволемии, связанной с задержкой натрия и воды в организме. Иными словами, почка, работающая в режиме «переключения», осуществляет достаточную экскрецию воды и натрия лишь при повышенном уровне АД.
Одним из фундаментальных открытий последних лет является определение роли дисфункции эндотелия в развитии сердечно-сосудистой патологии и патогенезе артериальной гипертензии, в частности. Под эндотелиальной дисфункцией сегодня понимают потерю эндотелием способности регулировать тонус и толщину сосуда, управлять процессами коагуляции и фибринолиза, оказывать противовоспалительное действие. В определенных ситуациях – при ишемии, гипоксии, повышении АД, имеет место снижение клетками эндотелия выработки NO и других факторов вазодилатации и усиление синтеза вазоконстрикторных субстанций – эндотелина, тромбоксана и др., т. е. развивается дисфункция эндотелия. При этом соответственно усиливаются процессы пролиферации, коагуляции, воспаления. Установлено, что важнейшим фактором эндотелиальной дисфункции является хроническая активация РААС, реализующая свои влияния с помощью главного эффекторного гормона ангиотензина II.
Таким образом, существующие патогенетические концепции артериальной гипертензии, по сути, объединяют все имеющиеся теории и во многом объясняют гетерогенность природы и разнообразие клинических форм этого заболевания. Согласно современным представлениям, артериальная гипертензия является результатом взаимодействия наследственных факторов, предрасполагающих к развитию заболевания и различных внешних воздействий, реализующих такую возможность.
Источник
Гипертоническая
болезнь –
артериальная гипертензия неизвестной
этиологии. Диагноз гипертонической
болезни (эссенциальной, первичной АГ)
устанавливают методом исключения
вторичных (симптоматических) артериальных
гипертензий.
Определение
«эссенциальная» означает, что стойко
повышенное АД при гипертонической
болезни составляет сущность этой
артериальной гипертензии. Каких-либо
изменений в других органах, которые
могли бы привести к артериальной
гипертензии, при обычном обследовании
не находят.
Классические
концепции этиологии и патогенеза
эссенциальной АГ включают нейрогенную
теорию Г.Ф.Ланга-А.Л.Мясникова, мембранную
Ю.В.Постнова-С.Н.Орлова, объемно-солевую
А.Гайтона, церебро-ишемическую Диккинсона.
Нейрогенная
теория Г.Ф.Ланга – А.Л.Мясникова.
Согласно концепции, разработанной Г.Ф.
Лангом и А.Л. Мясниковым, «нервное
перенапряжение при гипертонической
болезни реализуется в расстройстве
трофики определенных мозговых структур,
управляющих АД. Об этом свидетельствуют
частые случаи развития первичной
гипертензии у людей «стрессовых»
профессий. Особое значение при этом
имеют отрицательные эмоции, в частности
эмоции, не
отреагированные в двигательном акте,
когда вся сила их патогенного воздействия
обрушивается на систему кровообращения.
На этом основании Г.Ф. Ланг назвал
гипертоническую болезнь «болезнью
неотреагированных эмоций».
Непосредственный
механизм повышения АД связан с
возникновением очага
застойного возбуждения (патологической
доминанты, по терминологии А.А. Ухтомского)
вегетативных центров головного мозга,
в первую очередь сосудодвигательного
центра.
Стресс вызывает
активацию гипофизарно-надпочечниковой
и симпатоадреналовой системы с последующим
включением прессорного механизма РААС.
Это приводит к увеличению сердечного
выброса (МОК) при неизменной или слегка
сниженной величине ОПСС.
Рассмотренные
процессы формируются на первой
стадии гипертонической болезни,
которая называется транзиторной
(преходящей) и клинически характеризуется
непродолжительными эпизодами повышения
АД.
На второй стадии
гипертонической болезни (стадия
стабильной АГ) увеличение АД и местного
кровотока приводит к увеличению
сосудистого тонуса и генерализованному
спазму артериол (повышению ОПСС),
направленному на приведение в соответствие
местного кровотока и потребности в нем
тканей.
В происхождении
стабильного повышения тонуса сосудов
имеют значение формирующиеся в этой
стадии «порочные
круги» (рис.
1): барорецепторный, почечный (с участием
ЮГА), гипофизарно-надпочечниковый и
сосудистый (повышение чувствительности
стенки сосудов к катехоламинам).
В результате
повышения АД развивается парабиоз
барорецепторов сосудов и выпадает их
тормозной контроль над нейронами
сосудодвигательного центра. В итоге
тонус сосудов повышается еще сильнее.
Спазм сосудов
приводит к гипоксии ЮГА почек и активации
РААС.
В свою очередь,
ишемическая стимуляция аденогипофиза
реализуется в секреции АКТГ и,
следовательно, повышении содержания в
крови гормонов коры надпочечников
(минерало- и глюкокортикоидов). Поэтому
высокий тонус сосудов поддерживается
продолжительное время.
В механизме
гипертонии также играет роль повышение
чувствительности стенки сосудов к
катехоламинам, и даже небольшие дозы
адреналина оказывают выраженный
вазоконстрикторный эффект.
Таким образом,
определяющую роль в патогенезе АГ играют
изменения нейрогуморальной регуляции
сосудистого тонуса.
Конечным звеном
этого патологического процесса является
изменение функциональной активности
ионотранспортирующих систем плазматической
мембраны, что ведет к перегрузке
гладкомышечных клеток ионами Cа2+
и патологическому повышению тонуса
кровеносных сосудов. Такая концепция
патогенеза артериальной гипертензии
была выдвинута Ю.В.
Постновым и С.Н. Орловым,
которые назвали ее мембранной
концепцией.
Объемно-солевая
теория А. Гайтона.
Согласно этой теории, в основе развития
эссенциальной АГ лежит ослабление
выделительной функции почек, которое
приводит к задержке в организме ионов
Na+ и воды, а, следовательно, к увеличению
ОЦК и МОК.
При этом повышение
АД играет компенсаторную роль –
необходимо для обеспечения адекватного
натрийуреза и диуреза.
В ответ на повышение
МОК, местные механизмы саморегуляции
кровотока вызывают миогенное сужение
артериол, результатом которого является
нормализация МОК за счет повышения ОПСС
и тем самым повышения АД. Увеличению
выраженности и стойкости этой
констрикторной реакции способствует
повышение реактивности сосудов вследствие
отека и аккумуляции ионов Na+ в их стенке.
Таким образом, с
течением времени «гипертензия
выброса» со
свойственным ей гиперкинетическим
типом изменений гемодинамики (увеличение
МОК при нормальном ОПСС) трансформируется
в «гипертензию
сопротивления»
с гипокинетическим гемодинамическим
профилем (увеличение ОПСС при нормальном
или сниженном МОК).
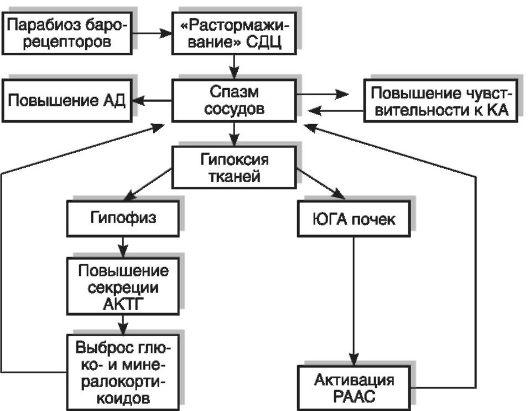
Рис. 1. «Порочные
круги» в
патогенезе гипертонической болезни:
АД — артериальное давление; АКТГ —
адренокортикотропный гормон; КА —
катехоламины; СДЦ — сосудодвигательный
центр; РААС — ренинангиотензин-альдостероновая
система; ЮГА — юкстагломерулярный
аппарат.
Церебро-ишемическая
теория Диккинсона. Уменьшение
объемной скорости кровотока в сосудах
головного мозга (атеросклероз, спазм
церебральных сосудов и т.д.) вызывает
ишемию мозга. Реакция на ишемию головного
мозга начинается с хеморецепторов,
расположенных в ЦНС, что приводит к
активации прессорного отдела
сосудодвигательного центра, сильной
симпатической вазоконстрикции,
стимуляции сердечной деятельности.
Увеличение системного АД позволяет в
определенной степени улучшить
кровоснабжение мозга, однако поддержание
стабильно высокого АД не может
осуществляться только за счет спазма
сосудов. Ишемия ЦНС, по-видимому, является
только инициирующим звеном АГ.
Соседние файлы в предмете [НЕСОРТИРОВАННОЕ]
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
Источник
